
Minding Your Binding: Plasma Protein Binding Potential Study Now Available at Our US Labs
While liver and intestine are important in ADME, behavior in blood is also crucial. When an orally administered drug or other xenobiotic enters the body, typically it is absorbed in the gut, enters the blood for systemic circulation then undergoes biotransformation in the liver, kidney and/or other organs, and is finally excreted in the urine or bile. Intravenous administration follows a similar path, exclusive of the gut. Binding with proteins in plasma can impact a drug’s distribution and clearance, potentially altering its efficacy and toxicity. Accurately measuring this binding is critical to understanding pharmacokinetics, as well as potential for drug-drug interactions (DDI).
As of April 15th, 2019 XenoTech now offers a definitive Plasma Protein Binding (PPB) study, carried out on our Kansas City campus, in addition to the PPB services available through our partner at the Tokai Drug Development Solution Center in Japan, to help customers paint a better picture of test compound pharmacokinetics that will impact preclinical and clinical study design considerations.
Features of the study include:
- Preliminary evaluations of stability, non-specific binding potential, and time to equilibrium
- Main experiment to accurately determine fraction unbound (fu) in plasma using rapid equilibrium dialysis (RED)
- Full report detailing methods, data, and results/conclusions
The free drug theory describes the common understanding that, in the absence of energy-dependent processes (such as uptake/efflux transporters and maintenance of a pH gradient), an increase in plasma protein binding results in a decrease in unbound drug available to diffuse into tissue and act therapeutically at a target site[1]. High plasma protein binding (PPB) can extend a drug’s half-life due to decreased clearance; distribution and metabolism are likewise affected. Understanding these parameters can help you answer the question, “What dosage will be appropriate for clinical trials?”
Our study design can include up to five clinically relevant species to evaluate these variances and improve interpretation of preclinical pharmacokinetics and human dose prediction for clinical work. Interspecies variability in plasma concentration is of high importance; for example, a drug which is highly bound in humans may not be in mice. Plasma concentration can even vary between patients, resulting in reciprocal changes in free drug concentration. For instance, changes in plasma concentration can occur in pregnant women or patients with hyperthyroidism or liver impairment.
Protein binding could also be a contributing factor in DDI concerns. For example, phenytoin and valproic acid are medications prescribed to treat seizures. Phenytoin is 90% bound, and when co-administered with valproic acid a displacement effect may be precipitated, whereby bound phenytoin is blocked from protein binding sites and unbound concentration increases by 30-100%, leading to toxicity[2].
Fraction of unbound drug (fu) is denoted as counterpart to percent of bound drug, and is an important component of preclinical data. Factors determining fu are comprised of a drug’s affinity to a protein, concentration of the protein in the plasma, and concentration of a drug relative to protein. Definitive species comparisons of PPB can assist in the prediction of human unbound Cmax values from animal data to inform dose range selection for human trials. Furthermore, the unbound Cmax concentration in humans is needed to select concentrations for in vitro drug-drug interaction studies required by the regulatory agencies.
Neglecting to quantify the fu of a compound accurately with a definitive study can translate to misinformed dose determination. Even a small inaccuracy can mean a big difference if the drug has high PPB, which is categorized by a binding affinity of greater than 99%[3]. Knowing whether your drug is 98.9% or 99.8% bound can de-risk changes in free drug exposure, which can impact in vivo studies. For example, this could mean the difference between a 10 mg and 30 mg dose for first in-human trials.
The standard study designed at XenoTech for drug developers interested in PPB determines the bound/unbound ratio using Rapid Equilibrium Dialysis, and our partner lab in Tokai also employs two other methods of determining PPB:
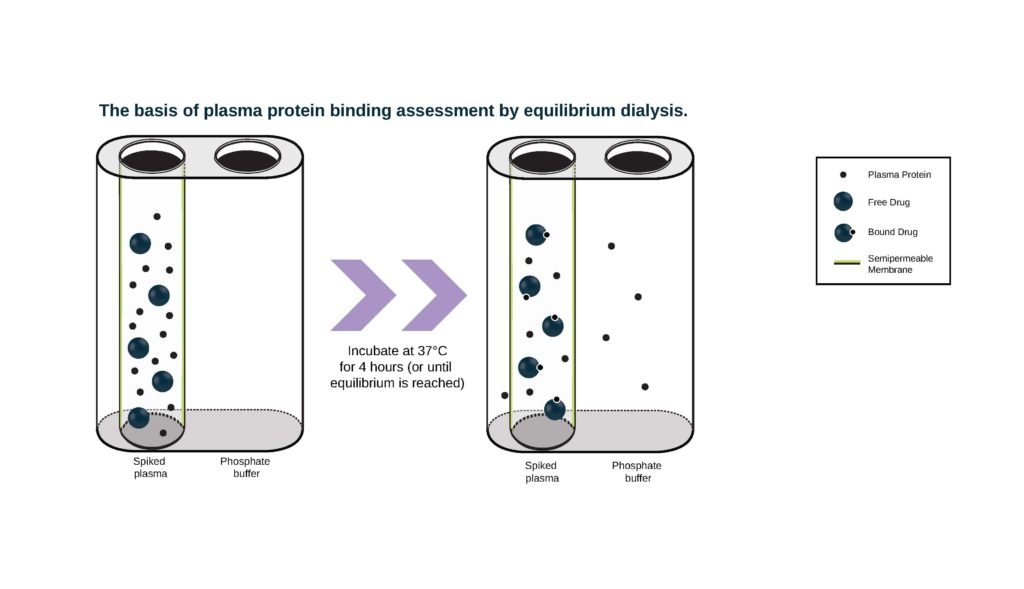
- Rapid Equilibrium Dialysis (RED) devices comprise a chamber containing plasma proteins separated from a chamber containing buffer by a dialysis membrane. We have different membrane options suited to different sized test article molecules: Molecular weight cutoff (MWCO) 8 KDa or 12 KDa. Drug is added to the chamber containing plasma and the chambers are incubated in a RED device plate at 37° C. Aliquots of buffer and plasma from both chambers are collected at appropriate time points and the amount of drug in each chamber is measured by either LSC (if compound is radiolabeled) or LC-MS/MS.
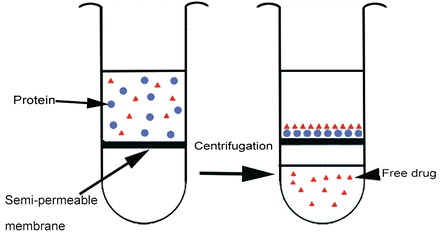
2. Ultrafiltration utilizes a semi-permeable horizontal membrane within a test vial. When protein and test article are loaded on top and centrifuged at 2000 g, protein-ligand complexes are trapped in the membrane, and the amount of free drug remaining in the protein-free filtrate is collected in the bottom chamber and measured.
3. Ultracentrifugation combines test compound with plasma and centrifuges the mixture at high force (e.g., 500,000 g) for long incubation times (up to 24 hours) at 37° C. The resulting stratification separates plasma proteins bound to drug into a pellet, and free drug in the supernatant is measured to determine fu.
Our standard design includes preliminary evaluations of compound stability, non-specific binding to the apparatus, and time to reach equilibrium to ensure the procedure is appropriate and optimized for the main experiment. ‘Stability’ typically refers to the interaction of test article with enzymes in the plasma or chemical degradation, which could result in decreased concentrations and make it difficult to accurately determine PPB. Non-specific binding and time to equilibrium are evaluated in buffer to evaluate the impact of non-specific binding to the apparatus and determine the appropriate incubation time that should be utilized for the evaluation of PPB in the main experiment.
Preliminary experiments are performed in duplicate at one test article concentration, measured at three predetermined, appropriate time points. Results are used to design the main experiment, in which bound/unbound fraction is measured at three different test article concentrations at one time point in triplicate. Human and major preclinical species can be evaluated and warfarin is included as the positive control.
| Experiment | (n) | TA concentrations | Time points | Results |
|---|---|---|---|---|
| Preliminary | n=2 | 1 | 3 (e.g. 4, 6, 24 hr) | Test article remaining Unbound drug remaining Equilibration time |
| Main | n=3 | 3 | 1, Determined in preliminary | Fraction of unbound Percent bound |
This service extends our wide variety of offerings designed to help drug developers move compounds with the highest likelihood of success through clinical testing. To get started today on your PPB study, contact a XenoTech Services Representative…
Learn more about:
[1] Bohnert, T., Gan, L. S., “Plasma Protein Binding: From Discovery to Development.” J. Pharm. Sci. 2013, 102, 2953– 2994. https://jpharmsci.org/article/S0022-3549(15)30955-2/pdf[2] Perucca, E, et al. “Interaction between Phenytoin and Valproic Acid: Plasma Protein Binding and Metabolic Effects.” Clinical Pharmacology and Therapeutics, U.S. National Library of Medicine, Dec. 1980, www.ncbi.nlm.nih.gov/pubmed/6777108.[3] Cohen, Lucinda H., and Deborah A. Nicoll-Griffith. “Plasma Protein Binding Methods in Drug Discovery and Development: Bioanalysis.” Encyclopedia of Drug Metabolism and Interactions, 2012, doi:10.1002/9780470921920.edm114.[4] Mukker J.K., Singh R.S.P., Derendorf H. (2016) Methodologies for Protein Binding Determination in Anti-infective Agents. In: Rotschafer J., Andes D., Rodvold K. (eds) Antibiotic Pharmacodynamics. Methods in Pharmacology and Toxicology. Humana Press, New York, NY
Contact Us
With Questions or Feedback, We’d love to hear your thoughts

Learn More
About XenoTech’s Contract Research Services
